
Research
De novo protein design in Foldit
I develop software tools for the crowdsourcing game Foldit, and set up protein design challenges for Foldit players. Then I analyze Foldit players’ designs, and test their proteins in the wet lab. The results of these tests are used to inform the next round of Foldit challenges and new design tools. As we improve the Foldit software, we challenge Foldit players with more difficult design tasks.
Foldit players have led us to some important conclusions about protein design and citizen science:
“Automated” protein design software is incomplete. Protein design is a complex problem, and researchers have built sophisticated design software that incorporates much of our knowledge about protein structure. Yet, even “automated” software typically requires some intervention from an expert protein engineer. It is unclear how much knowledge is encoded in our software, and how much we rely on the implicit knowledge and expertise of the user. Citizen scientists, by using our software without expertise, offer a rigorous test of the knowledge underlying the software. Citizen scientists playing Foldit have revealed implicit assumptions in our protein design methods, and have discovered flaws in our protein design score function. We addressed these flaws and assumptions in Foldit, and players can now successfully design new proteins from scratch.
Citizen scientists are creative. Because protein structures have many degrees of freedom, the number of possible protein structures is astronomically large. Several billion years of natural evolution has explored only a tiny fraction of possible protein folds. So there is considerable room for creativity in designing new protein folds. When challenged to design new proteins from scratch, Foldit players explore a greater variety of folds than researchers or computational methods – including protein folds that don’t exist in nature.
Our results suggest that citizen science might be useful for testing scientific models in general, and that crowdsourcing is especially well-suited for creative tasks.
Model building in Foldit with electron density
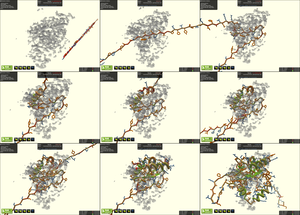
Structural biology depends heavily on accurate protein structures, which are usually based on x-ray crystallography and cryo-electron microscopy (cryo-EM) experiments. These experiments produce a high-resolution electron density map of the protein structure. Building a model into the electron density map is a critical part of structure determination, but can be labor-intensive and prone to error when carried out by a single individual.
We’ve added support for electron density maps in the protein folding game Foldit, allowing us to crowdsource model building to Foldit players. Although Foldit players lack formal training in protein structure, the Foldit energy function guides players toward realistic models that match the experimental data. Foldit players are adept at interpreting visual electron density maps, and – collectively – can produce high-quality, accurate models on par with those of structural biologists.